PERFILES
3
MÉTODOS COMPUTACIONALES
PARA EL DESARROLLO E INVESTIGACIÓN
DE LA NANOCIENCIA
Palabras Clave: Nanociencia, DFT, ab initio, nanomateriales, estructura electrónica.
Keywords: Nanosciences, DFT, ab initio, nanomaterials, electronic conguration.
Research and development of nanosciencie may be considered as a real revolution in several elds of knowledge and industry, it is based
on chemistry and physics theory. Nowadays with the overcome of more powerful and new computers one can create theoretical models
for the study of nanosytems, these are known as computational methods and can be applied at nanometric scale. The most used methods
are the electronic structure, and the principal methodology is the Density Functional Theory (DFT). These tools are a support for research
and allow us to obtain information that is not easy to retrieve with experimental methods. Quantum-mechanics calculus can be done with
computational packages of different kinds developed by research groups and Universities.
Cristian Vacacela,
1,3
Javier Torres,
2,3
Robert Cazar,
1,3
Dennis Cazar.
1,3
1
Facultad de Ciencias – Escuela Superior Politécnica de Chimborazo,
Panamericana Sur 1 ½ km, Riobamba-Ecuador.
2
Colegio de Ciencias-El Politécnico Universidad San Francisco de Quito,
Diego de Robles y Vía Interoceánica, Quito-Ecuador.
3
Grupo Ecuatoriano para el Estudio Experimental y Teórico de Nanosistemas, GETNano.
Autor para correspondencia: cristianvg7@gmail.com
Fecha de recepción: 1 de abril de 2013 - Fecha de aceptación: 25 de julio de 2013
Imagen ilustrativa: Fullerene nanogears. Fuente: Wikimedia commons
El desarrollo e investigación de la nanociencia ha constituido una verdadera revolución en diferentes campos del conocimiento y la industria,
se la puede llevar a cabo con las bases teóricas de la física y química. Actualmente con la aparición de nuevos y más potentes ordenadores
se pueden crear modelos teóricos para el estudio de los denominados nanosistemas, a esto se conoce como métodos computacionales
aplicados a escala de nanómetros. Los métodos más utilizados son los de estructura electrónica, y la principal metodología es la Teoría
del Funcional de la Densidad (DFT). Estas herramientas son un soporte para la investigación y permiten obtener información que no es
posible conseguir con métodos experimentales. Los cálculos cuanto-mecánicos se pueden realizar en paquetes computacionales de diversa
índole desarrollados por centros de investigación y universidades.

PERFILES
4
1. INTRODUCCIÓN
El estudio de la materia a escala atómica ha sido
afrontado por varias ramas de la ciencia desde los
principios del siglo XX, no puede denominarse
química, física o biología dado que los cientícos
están estudiando un campo dimensional muy pe-
queño que abarca átomos y moléculas, que son
los “ladrillos” con los cuales se construye la ma-
teria a nivel macroscópico. La nanociencia es un
estudio puramente teórico que trata de compren-
der y predecir el comportamiento de sistemas
formados por átomos y moléculas. El signicado
de “nano” es una dimensión: 10 elevado a -9, es
decir un billonésimo de metro. [1]
El desarrollo tecnológico ha visto en los resulta-
dos de la nanociencia la oportunidad de redenir
el modelo de proyectación de dispositivos y ma-
teriales, proponiendo un esquema denominado
bottom-up, es decir construir un determinado
elemento partiendo desde su composición a ni-
vel atómico; la aplicación de los descubrimientos
hechos en nanociencias es lo que se conoce como
nanotecnología. [2, 3]
El estudio de la nanociencia conlleva el manejo
de teorías a nivel atómico, es decir física y quí-
mica cuántica, a la par de métodos numéricos
que permitan resolver las complejas ecuaciones
que describen los nanosistemas. Básicamente los
métodos numéricos son herramientas de cálculo
que emplean ordenadores (HPCs, clústers y grids)
para obtener soluciones numéricas que permitan
predecir el comportamiento de sistemas comple-
jos [4], en este contexto se pueden diferencias dos
grandes conjuntos los métodos de mecánica mo-
lecular que emplean las leyes de la mecánica clá-
sica y los métodos de estructura electrónica que
emplean la mecánica cuántica [5, 6, 7]. La elec-
ción de uno o del otro depende de una serie de
factores tales como el tamaño del sistema, el tipo
de información y sobre todo el tiempo de cálculo
requerido, en este último los cálculos estructura
electrónica son más costosos que los de mecánica
molecular, sin embargo, en la nanociencia son ne-
cesarios para el estudio de los electrones de valen-
cia, los cuales describen como se unen los átomos
y por ende como se forma la materia. [8]
En Ecuador se están formando grupos de pro-
fesionales, docentes, investigadores y estudian-
tes que realizan trabajos de investigación en na-
nociencias, en particular GETNano [21] grupo
conformado por varias universidades de la nación
lleva adelante proyectos nalizados al estudio y
divulgación de la nanociencia a nivel nacional,
el presente artículo se basa en un trabajo de in-
vestigación conjunto que ilustra el proceso de
investigación en nanociencias y su aplicación en
nanotecnología.
2. MÉTODOS Y TÉCNICAS
La nanociencia es el estudio teórico de la materia a escala manométrica
(del orden de 10
-9
m.), la cual describe el comportamiento de sistemas
multielectrónicos, en particular, la conguración geométrica que corres-
ponde al estado de mínima energía. A niveles cuanto-mecánicos esto
signica resolver la ecuación de Schrödinger, denida así [9]:
Donde, E es la energía, Ψ es la función de onda del sistema y H^ es el
operador Hamiltoniano no relativista independiente del tiempo, la solu-
ción analítica de la ecuación (1) se obtiene solo en muy pocos casos y en
otros se deben realizar aproximaciones que nos llevan a soluciones nu-
méricas. Esencialmente en la química cuántica, para resolver la ecuación
de Schrödinger se deben separar el movimiento nuclear del electrónico,
de acuerdo con la aproximación Born-Oppenheimer [10]. Debido a que
el núcleo tiene mayor masa que los electrones, se puede considerar que
su movimiento es lento en comparación a estos y suponer que los elec-
trones se mueven en un campo creado por núcleos jos. Entonces, la
ecuación (1) es redenida como sigue [6, 10]:
Donde Ψ_e es la función de onda electrónica que depende explícita-
mente de las coordenadas de los electrones (r) y de las coordenadas
paramétricas de los núcleos (R), E_e representa la energía del sistema
electrónico para una determinada distribución de los núcleos y H^ es el
Hamiltoniano electrónico que incluye la energía cinética de los electro-
nes y la energía electrostática de atracción (núcleo-electrón) y repulsión
(electrón-electrón) [5, 6]. La solución de la ecuación (2), nos da la geo-
metría que tendría un sistema si los núcleos fuesen estacionarios (Figura
2.). Si se considera el movimiento de vibración de los núcleos, la solu-
ción de la ecuación (2) es una aproximación a la estructura promedio del
sistema en el estado fundamental. La introducción de esta consideración
hace que los métodos cuánticos se vuelvan más complicados y requie-
ran mayor tiempo de cálculo; existen dos métodos fundamentales que
estudian este problema, estos son los métodos ab initio y los métodos
semiempíricos. [5, 6]
Figura 1. Logo de la IBM formado por 35 átomos y visualizado con el uso
de un microscopio de fuerza atómica [1].
(1)
(2)
PERFILES
5
Los métodos ab initio se derivan directamente de los principios teóricos
y no incluyen ningún parámetro experimental en sus ecuaciones, esto no
implica que la solución sea exacta sino aproximada, el más simple de este
tipo de cálculo es el método Hartree-Fock [11]. En cambio, los métodos
semiempíricos obtienen algunos parámetros de datos experimentales,
pudiendo ser empleados para tratar sistemas de muchos cuerpos donde
el formalismo de Hartree-Fock es computacionalmente muy costoso.
Los métodos semiempíricos más antiguos, son el Hückel y Hückel Ex-
tendido, y los más difundidos son los métodos: MNDO (The Modied
Neglect of Diatomic Overlap), MINDO (There are three Modied In-
termediate Neglect of Differential Overlap), AM1 (Austin Model One)
y PM3 (Parameterization Method Three). [5]
2.1 Teoría del Funcional de la Densidad (DFT)
Actualmente la Teoría del Funcional de la Densidad es la más utiliza-
da en nanociencia [12], dado que la energía total del sistema se puede
expresar en términos de la densidad total ρen lugar de la función de
ondaΨ, convirtiéndose en una alternativa a la solución de la ecuación
de Schrödinger [12]. Además, esta teoría puede ser considerada como
un método ab initio, por ser en principio una teoría exacta, pero es tam-
bién un método semiempírico ya que se aplica de forma aproximada [5].
Esencialmente la DFT, depende solo de 3 variables a diferencia de la
función de onda electrónica que depende de 3N variables (N = número
de partículas), conceptualmente se basa en el modelo de Thomas-Fermi
[13], pero su base teórica fue dada por Hohenberg y Kohn en 1964
[14], quienes formularon dos aspectos importantes, (i) todos los obser-
vables de un sistema de N-cuerpos son determinados por la densidad
electrónica en el estado fundamental y (ii) la densidad electrónica es un
funcional de la energía del sistema [14]. Sin embargo, el desarrollo más
importante fue dado por Kohn y Sham en 1965 [15], quienes probaron
que la energía electrónica del estado fundamental E0 es una función de
[ρ0] y se dene así:
Donde, E
0
=E[ρ
0
] es la energía total del sistema
del estado fundamental, F[ρ
0
] representa el fun-
cional universal que contiene a la energía cinética
T[p0] y Vee[p0]es la interacción de repulsión elec-
trón-electrón, Ven[p0]=∫drp0 (r)v(r) es la interac-
ción de atracción electrón-núcleo, siendo v(r) el
potencial externo, es decir, la función de energía
potencial de atracción nuclear para un electrón
localizado en el punto r. Así, mientras Ven[p] es
conocido, se desconoce la forma exacta del fun-
cional F[p] [15]. La ecuación (4), no proporciona
una vía practica para calcular E0 a partir de p0, ya
que se deben hacer aproximaciones sobre F[p].
Dada esta necesidad, se han desarrollado funcio-
nales híbridos como el B3LYP [16] y funciona-
les doblemente híbridos como el B97D [17], la
diferencia entre el primero y el segundo es que
B97D toma en cuenta la energía de intercambio y
correlación EXC [p], y B3LYP no lo hace [17], de
esta manera el funcional B97D obtienen mejores
aproximaciones, adicional a esto se pueden mejo-
rar los cálculos añadiendo Basis Sets. [18]
2.2 Paquetes Computacionales
Una vez denido el nivel de teoría a emplear, se
pueden calcular las propiedades: estructurales,
electrónicas, vibracionales y termodinámicas, la
base para obtener todos los observables anterio-
res es empezar por las propiedades estructurales,
esto se conoce como optimización geométrica
(OPT), que consiste en encontrar la posición
relativa de cada átomo con respecto a los otros,
respetando el criterio de mínima energía, descar-
tando los mínimos locales para encontrar el míni-
mo global. El paquete de cálculo computacional
más importante para realizar estas tareas es Gaus-
sian09 [46] y los software para visualizar gráca-
mente los resultados pueden ser GausView05 y
Moldraw, pero los resultados numéricos son los
que ofrece la mayor cantidad de información y
se pueden vericar en cualquier editor de texto;
básicamente consisten en tener un archivo input
(le.in) con todas las coordenadas de los átomos
(x, y, z), y el tipo de cálculo a realizar, posterior-
mente se obtiene un archivo output (le.log) con
el resultado numérico del cálculo realizado.
3. RESULTADOS Y DISCUSIONES
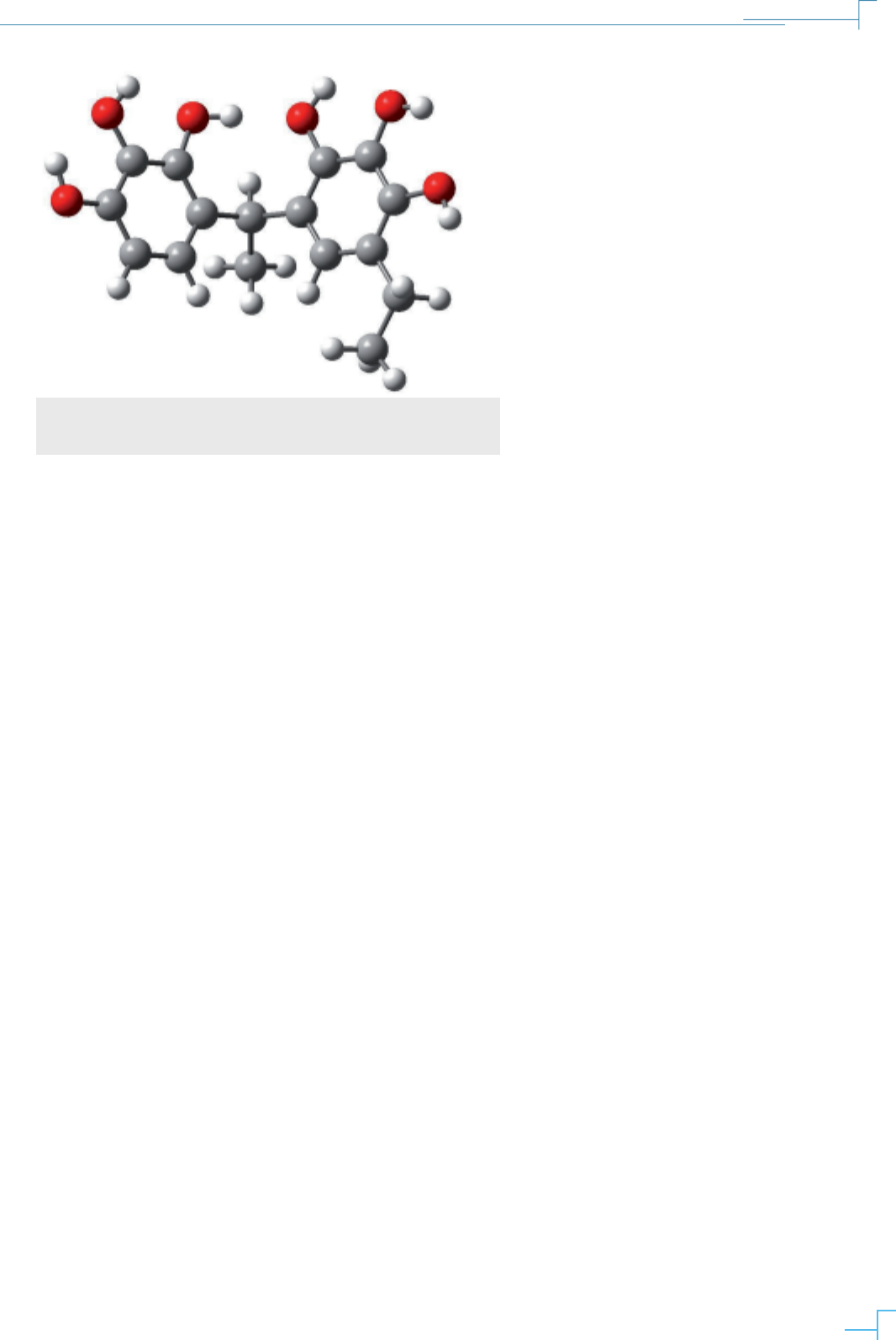
En la gura 3., se muestran los resultados de la
optimización de la geometría del R-Pyg[2]Ar (R
= metil) donde se pueden apreciar los mínimos
locales y el mínimo global, este último correspon-
de a la estructura de mínima energía y por tanto la
estructura más estable.
Figura 2. Optimización geométrica de dos unidades pirogalol, orientación
aproximada de los grupos hidroxilo y del grupo R = metil, realizado con
Gaussian09 y visualizado con GaussView5.0.
EE
000
=
[]
ρ
EE Fdrrvr
00 00
=
[]
=
[]
+
()()
∫
ρρρ
ETVeeVen
ρρ
ρρ
00
00
[]
=
[]
+
[]
+
[]
(3)
(4)
(5)
De donde E
0
=E[ρ
0
] se expresa como:

PERFILES
6
Figura 1. Logo de la IBM formado por 35 átomos y
visualizado con el uso de un microscopio de fuerza
atómica [1].
Los resultados obtenidos en el archivo output son
de vital importancia de acuerdo al tipo de estudio
que se realice, sin embargo, los métodos compu-
tacionales no permiten dar conclusiones, su ob-
jetivo principal es aportar la mayor cantidad de
información y ser un complemento a la investi-
gación experimental. Aunque relativamente estas
herramientas son propias de la física y química
computacional se pueden emplear para la inves-
tigación y desarrollo de la nanociencia, dado que,
teóricamente se puede predecir la formación de
los denominados nanomateriales o fundamentar
un hecho experimental poco estudiado como lo
son los R-Pirogalol[4]arenos (R = grupo aromá-
tico/alifático), y por ejemplo sus respectivas apli-
caciones como posibles nanoestructuras de alma-
cenamiento de moléculas de pequeña y mediana
dimensión, especícamente el H
2
[19].
4. CONCLUSIÓN
La rápida evolución de los ordenadores y de los
instrumentos para “ver” a escala atómica (micros-
copios de efecto túnel, microscopios electrónicos
de barrido, etc.) y el notable desarrollo de la física
y química computacional han propiciado la incor-
poración de modelos teóricos para el tratamiento
de todos aquellos problemas relacionados con la
geometría y la energía de un determinado sistema.
Los métodos computacionales más óptimos para
el desarrollo de la nanociencia son los métodos de
estructura electrónica, ya que permiten modelar y
simular el comportamiento de átomos y molécu-
las a escala de nanométrica, pudiendo construir
nuevos materiales partiendo desde su estructura
molecular y dejando este reto a la síntesis experi-
mental, sin embargo su aporte no queda ahí, tam-
bién, ayudan a obtener de información que no es
asequible experimentalmente.
Además son un soporte para la investigación ex-
perimental, aunque el limitante de los métodos de
estructura electrónica sean el recurso computa-
cional y el tiempo de cálculo requerido, los resul-
tados son muy satisfactorios. [20].
La nanociencia en el Ecuador puede desarrollar-
se sin problemas a niveles equivalentes de países
más desarrollados, ya que el bajo costo y las altas
prestaciones de los computadores comerciales hacen posible la imple-
mentación de clústeres
de alta potencia de cálculo a un costo asequible a los presupuestos de
Universidades e Institutos de Investigación, lo que más se necesita es
talento humano con deseo y curiosidad, formados sólidamente en cien-
cias básicas (Física, Química, Biología) que deseen afrontar el reto de
comprender los fenómenos naturales a niveles nanoscópicos.
AGRADECIMIENTOS
A Dios, al Grupo Ecuatoriano para el Estudio Experimental y Teóri-
co de Nanosistemas GETNano, a la Escuela Superior Politécnica de
Chimborazo, al Consorcio Ecuatoriano para el Desarrollo de Internet
Avanzado CEDIA.

PERFILES
7
[1] RAMSDEN, J. (2005). “What is nanotechnology?”. (págs. 3-17). Craneld: Departament of Advance Materials .
[2] RANJAN, D., & TRIPATHI, A. (2009). “Computational Nanotechnology: an Assessment”. (págs. 233-241). Buca-
rest: Journal of Nanomaterials and Biostructures
[3] CAO, G.. “Nanostructures & Nanomaterials. Synthesis, Properties & Applications”.. London: Imperial College
Press. (2004) (págs. 135-138)
[4] GAO, B., JIANG, J., & LUO, Y. (2009). “Simulation of electronic structure of nanomaterials by central insertion
scheme”. (págs. 307-314). Chengdu: Phys. China
[5] YOUNG, D. (2001). Computational Chemistry: A practical Guide For Applyin Techniques to Real-Word Problems.
(págs. 42-58). New York: Wiley-Interscience
[6] LEVINE, I., Química Cuántica., España., Pearson Educación S. A.,. 2001. Pp. 467-572.
[7] FORESMAN, J. (1995). Exploring Chemistry with Electronic Structure Methods. (págs. 86-123). Pitsburgh: Gaus-
sian Inc.
[8] BRAUN, T., SCHUBERT, A., ZSINDELY, S., Nanoscience and Nanotechnology on the Balance., Elseiver Science
Ltd (Oxford)., Marzo 1997., Pp.321-325.
[9] SCHRODINGER, E. (December-1926). An Ondulatory Theory of the Mechanics of Atoms and Molecules . (págs.
1049-1070). Zurich, Switzerland: Physical Review.
[10] HANDY, N., & YAMAGUCHI, Y. (january-1986). The diagonal correction to the Born–Oppenheimer approxima-
tion: Its effect on the singlet–triplet splitting of CH2 and other molecular effects. (pág. 4). California : Journal of Chemical
Physics .
[11] FISCHER, C., Hartree-Fock method for Atoms, a Numerical Approach., New York., John Wiley and Sons, Inc.,
1977., Pp.317.
[12] KRYACHKO, E., LUDEÑA, E., Energy Density Functional Theory of Many-Electron System., The Netherlands.,
Kluwer Academic Publisher., 1990., Pp.850.
[13] DREIZLER, R., GROSS, E.,. Density Functional Theory., 1ª ed., Berlin., Editorial Springer-Verlag., 1991., Pp.302.
[14] JONES, R. (1989). “The density functional formalism, its applications and prospect.”. (págs. 55-61). Canberra: Re-
view of Modern Phys.
[15] KOHN, W., & SHAM, L. (November-1965). Self-Consistent Equations Including Exchange and Correlation
Effects. (págs. 1133-1138). California: Physical Review.
[16] BECKE, A., Density-Functional Exchange-Energy Approximation With Correct Asymptotic Behavior., Journal
Physical Review (Canada)., September 1988., Pp.3098-3100.
[17] GRIMME, S. (January-2006). Semiempirical hibrid density functional with perturbative second-order correlation.
(págs. 1-17). Münster, Germany: American Institute of Physical
[18] MCLEAN, A., & CHANDLER, G. (May-1980). Contracted Gaussian basis for molecular calculations/ Second row
atoms, Z=11-18. (págs. 5638-5648). San Jose, California: Journal Chem. Phys.
[19] URBINA, A., SALTOS, A., & TORRES, J. (December-2011). Estudio computacional B3LYP de la interacción del
hidrogeno molecular [H2] con rccc R-Pyg[4]arenos [R = metil, uor] funcionalizados con Li+. (págs. 30-34). Quito: Avances:
En Ciencias e Ingenieria.
[20] WANG, L. (October-2009). Novel Computational Methods for Nanostructure Electronic Structure Calculations.
(págs. 19-39). Berkeley, California: Annu. Rev. Phys. Chem.
[21] GETNano Grupo para el Estudio Experimental y Teórico de Nanosistemas www.getnano.espoch.edu.ec